 and IRC with Gaussian09")
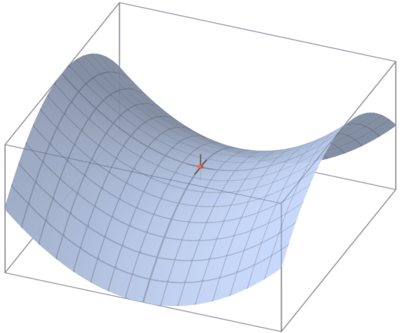
Theoretical evaluation of a reaction mechanism is all about finding the right transition states (TS) but there are no guarantees within the available methods to actually find the one we need. Chemical intuition in the proposal of a mechanism is paramount. Let’s remember that a TS is a critical point on a Potential Energy Surface (PES) that is a minimum in every dimension but one. For a PES with more than two degrees of freedom, a hyper-surface, envisioning the location of a TS is a bit tricky, in the case of a three dimensional PES (two degrees of freedom) the saddle point constitutes the location of the TS as depicted in figure 1 by a section of a revolution hyperboloid.

The following procedure considers gas phase calculations. Nevertheless, the use of the SCRF keyword activates the implicit solvent calculation of choice in order to evaluate to some degree the solvent influence on the reaction energetics at different temperatures with the use of the temperature keyword.
The first step consists of a high level optimization of all minimums involved, such as reagents, products and intermediates, with a subsequent frequency analysis that includes no imaginary eigenvalues.
In order to find the structures of the transition states we use in Gaussian the Synchronous Transit-guided Quasi-Newton method [1] through the keywords QST2 or QST3. In the former case, coordinates for the reagents and products are needed as input; for the latter keyword, coordinates for the TS structure guess is needed also.
QST2)
%chk=file.chk
%nprocshared=n
%mem=nGB
#p opt=(qst2,redundant) m062x/6-31++G(d,p) freq=noraman Temperature=373.15 SCRF=(Solvent=Water)
Title card for reagents
Q M
Cartesian Coordinates for reagents
—blank line—
Title card for products
Q M
Cartesian Coordinates for products
—blank line—
QST3)
%chk=file.chk
%nprocshared=n
%mem=nGB
#p opt=(qst3,redundant) m062x/6-31++G(d,p) freq=noraman Temperature=373.15 SCRF=(Solvent=Water)
Title Card for reagents
Q M
Cartesian Coordinates for reagents
—blank line—
Title card for products
Q M
Cartesian Coordinates for products
—blank line–
Title card for TS
Q M
Cartesian Coordinates for TS
—blank line—
NOTE: It is fundamental that the numbering order is kept constant throughout the molecular specifications of all two, or three, input structures. Hence, I recommend to build a set of molecules, save their structure, and then modified the coordinates on the same file to produce the following structure, that way the number for every atom will remain the same for every step.
As I wrote above, there are no guarantees of finding the right TS so many attempts are probably needed. Once we have the optimized structures for all the species involved in our mechanistic proposal we can plot their energies very simply with MS Excel the way we’ve previously described in this blog (reblogged from eutactic.wordpress.com)
Once we’ve succeeded in finding the structure of our TS we may run an Internal Reaction Coordinate (IRC) calculation. This calculation will connect the TS structure to those of the products and the reagents. Initial constant forces are required and these are commonly retrieved from the TS calculation checkpoint file through the RCFC keyword.
%chk=QST3_2p.chk
%nprocshared=8
#p m062x/6-31++G(d,p) IRC=(Maxpoints=50,RCFC,phase=(2,1))Temperature=373.15 SCRF=(Solvent=Water) geom=allcheck
Title Card
Q M
—blank line—
Finally, the IRC path can be visualized with GaussView from the Results menu. A successful IRC will link both structures along a single reaction coordinate proving that both reagents and products are linked by the obtained TS.
Hat tip to Howard Diaz who has become quite skillful in calculating these mechanisms as proven by his recent poster at the XII RMFQT a couple of weeks back. And as usual thanks to everyone who reads, comments, likes, recommends, rates and shares my silly little posts.

It’s a little misprint. In QST3 input you put: opt=(qst2…
Nevertheless. Already for a long time I wanted to say thank you for your blog, you do a great and important work! 🙂
Thanks for noticing the typo! I will fix it right away. Also thank you for your kind words about this blog 🙂
Have a nice day!
I am trying to get a transition state having a complex molecular system with 4 H-bond. the The groups invoved for interaction are C=O and NH2. The distance between them is 4.5 angstrom initially. I got a transition state from it with 48 kCalmol(-). It seems that I have missed a transition state in between. What procedure should I take to get the transition state in between the H bonded initial complex and the proton shuttling TS?
Thanks in support of sharing such a pleasant idea, paragraph is
good, thats why i have read it fully
Hola, estoy intentado proponer un estado de transición que es básicamente una disociación para el (CH3)3CC(O)OO—-NO2 pero cuando obtengo el TS la frecuencia negativa que encuentro es muy pequeña así que necesito mejorar esa frecuencia. Los keywords que estoy usando son
# opt=(calcfc,tight,ts,noeigentest) freq ub3lyp/6-31++g(d,p)
Me podría dar algunas recomendaciones que me ayuden a mejorar el cálculo?
Muchas gracias.
Hola Diana!
A que te refieres con muy pequeña? Ni en valor ni en intensidad (IR o RAMAN) necesitas que sea “grande” si corresponde al estado vibaracional que conecte tus reactivos con tus productos.
Por lo que veo tu nivel de teoría es adecuado, si bien el uso de b3lyp pudiera acaso ser cuestionable aunque nosotros hemos obtenidos buenos resultados (reproducciones experimentales) con dicho funcional.
Saludos y estoy al pendiente
La frecuencia IR es -54.23, lo que quiero observar es la separación entre el O—N del sistema así que digamos que no necesariamente que sea grande pero que sea la frecuencia para este vibración. Y bueno precisamente estoy buscando este estado de transición porque quiero compararlo con lo obtenido experimentalmente. No se si queda claro?
Pues si esa frecuencia de -54.23 corresponde a la separación O-N que buscas pues ya terminaste! O estoy entendiendo mal?
Excellent article. Keep writing such kind of information on your blog.
Im really impressed by it.
Hello there, You’ve performed an incredible job. I will
definitely digg it and in my opinion recommend to my friends.
I am confident they will be benefited from this website.
Buenas Joaquin:
Encontre un TS a nivel HF/3-21G* con una frecuencia imaginaria de -186 cm-1 y el calculo del IRC me da perfecto. Sin embargo al querer reoptimizar esa estructura usando M062X/6-311+G** me genera una estructura con una frecuencia negativa muy pequeña -39 cm-1 y el IRC solo me da dos pasos y se corta. No estoy segura si en realidad es un TS.
‘m working in IRC calculation in Gaussian 09. The job was terminated with the following error message. How can I resolve it?
The error message: Maximum number of corrector steps exceded.
Error termination via Lnk1e in g09/l123.exe
i am very new to gaussian.can u please help me for IRC calcualtion???
Hi
If it possible one exaple about QST2 and QST3 and RIC
Thanks a lot
Hi thanks for the useful information.
I am trying to model a complex reaction and found the optimised reagents and products (from opt jobs) to be higher in energy than the end structures from IRC curves. I think this is a rather odd problem as I think they should be lower in energy if anything. Is there an immediate reason why? Freq jobs show no imaginary frequencies for the optimised reagents/products, and indeed, the opt job went to completion – but I understand they might have been optimised at a higher minimum.
The transition states were aquired with opt=qst3 and show only one imaginary frequency. The IRCs do show reagent/product formation for reverse and forward directions, but of course the bond lengths and angles aren’t exact which is a little expected. I’ve been using the B3LYP/6-311++G(d,p) level as this showed the least sharing of MOs due to lack of diffuse functions on lesser basis sets.
The root line for the optimised reagents is:
#n B3LYP/6-311++G(d,p) Opt Freq
The root line for the transition states is:
#n B3LYP/6-311++G(d,p) Opt=QST3 Freq
The root line for the IRCs is:
# B3LYP/6-311++G(d,p) irc=(CalcFC,forward,maxpoints=79,stepsize=10) geom=check
I used 79 max points as this was the maximum path steps found for the run when I initially set 200 maxpoints.
Any help would be most appreciated,
Leighton
Hi Leighton
We have come across this problem too. It is probably due to a complex potential energy surface. Unfortunately the only advice I can give you is to play -systematically- a bit with geometries and levels of theory. Using the NoEigenTest keyword might help you but it will consume more computing time.
I hope this helps, let me know!
Hi Joaquin,
Unfortunately, the NoEigenTest did not change the situtation – although the good news is that the NoEigenTest ran fine and jobs went to completion! Thanks for the suggestion all the more.
So now another question along similar lines if I may…I would like to calculate the energy surface, starting with the saddle point and then moving down the IRCs steepest descent forwards and reverse to the local minima (probably done over many jobs). How do I do this? I can only find Scan keywords where the only examples provided are dihedral rotations, but not the generation of a saddle point surface. Can you recommend a keyword in Gaussian 09 that can achieve this please?
I would like to specifically refer to the type of surface by referencing Figure 1 in the following freely accessible article: http://chem.wayne.edu/schlegel/Pub_folder/265.pdf
Best regards,
Leighton
Kudos
I use your page for tutoring students…
Manythanks…
k
Wow! Thanks a lot!
I’m very glad to know the blog is useful for your lessons, I’ll try to keep it up.
Best wishes
Hi Dr. Joaquin,
Thanks a lot for your useful blog.
please, I want to know how can I convert temperature and pressure in G03W, we know that the default temperature is 298.15K and pressure 1 atm. I need to make my calculations at another conditions (e. g. T=0K). my calculations are not frequencies or thermochemistry properties, they are energies for a system.
I used
# … Temperature=0K
but it is failed
Hello,
The idea of pressure and temperature for a single molecule in a vacuum lacks any physical meaning. The Temperature keyword is used for the thermochemistry step but it cannot be used for the energy of a single molecule.
I hope this helps
Hello,
I’ve been trying to find the TS barrier energies associated with a linear N—-O bond breakages. I’ve been using TS with noeigentest and have had one of two results:
A TS showing the suspected product molecule (bond unbroken), typically with neg freqs (1-3) showing rotational degrees of freedom.
A TS showing an infinitely separated complex, usually ending in many errors.
TS search is a frustrating (and rewarding) venture to say the least. Nucleophilic substitutions are pretty simple to find, but bond breakage has been giving me troubles. Any tips for moving forward? I haven’t had much luck with typical hybrid density functionals (b3lyp) with reasonably sized basis sets.
Thanks for any advice,
Daniel
Hello Daniel,
“S search is a frustrating (and rewarding) venture to say the least” you said it! It is indeed a bit of an art form.
It seems you are on the right track but maybe changing functionals would go a long way. Try using M06-2x instead of b3lyp, that way you’ll get the long range interaction right. Also try the QSTn methods at least to a first approximation to the geometry of your TS.
I hope this helps. Have a nice day!
Hi, I’ve tried to run this calculation and i got this error ” Maximum number of corrector steps exceded.”.
Could you help me?
Hi DrJoaquin
what can be done if an IRC run in g09 goes in the same direction for both the reverse and forward keywords?I try with keyword phase but see the
same problem.what time does use keyword phase? which had atoms chosen? I try with atoms that formed the new bond between two molecules. is it correct? please guide me.
Hi Dr Joaquin many thanks for your good answers,please if you illustrate qst2 and irc with clear ep.
Hola, Joaquín. En primer lugar felicitarte por el blog, pero tengo algún problema con el cálculo. Estoy usando la siguiente línia de comandos:
#P b3lyp/6-31+G** Opt=(qst2,redundant) geom=allcheck gfprint gfinput gfoldprint SCRF=(PCM,SOLVENT=WATER)
Pero me da el siguiente error:
Number 0
Base 40448
End 65536
End1 65536
Wr Pntr 40448
Rd Pntr 40448
Length 25088
Error termination in NtrErr:
NtrErr Called from FileIO.
He leído por ahí que puede ser que falte memoria de cálculo, pero estoy usando 33 GB. Además, no me funciona con otras bases, ni tan siquiera con una hf/3-21G.
Tienes alguna idea de dónde puede estar el problema?
Muchas gracias
Posiblemente se trata de un error en la geometría, en tu línea de entrada dice que estás leyendo la geometría del archivo chk, eso es correcto?
La última línea del error indica que estás leyendo desde un archivo que no necesariamente existe.
Si en tu carro estás definiendo la creo metría de entrada elimina el comando geom=allcheck y con eso debería funcionar
Hola joaquin, buenas noches, felicitaciones por el blog, me ha sido util.
Estoy realizando un calculo IRC, cuando el proceso termina, me aparece este mensaje: this type of calculation cannot be archived. A que se debe esto?
Gracias
Hi Joaquin,
I’ve been trying to run some backwards and forwards IRC calculations and keep getting this error message “This type of calculation cannot be archived”. I’ve also encountered it while performing scan calculations. I am very unsure what it means and whether I can still use the results, as there doesn’t seem to be an actual error.
I would be great if you knew what it meant as I cannot seem to find much elsewhere about it. Your blog is a great source of information for learning the ins and outs of computational chemistry, thank you it has been very useful.
Emilie
Dear Dr. Joaquin
How are you doing? 🙂 It’s me, again. I have a question regarding the study of TS using the input “scan”. I was wondering if there’s a difference between doing a scan in gaseous phase and then optimizing the TS in a solvent and doing the scan in the solvent from the beginning.
I run a calculation using both ways and got a small difference in the data, but I’m not sure it’s a significant difference. I hope you can illustrate me in the matter 🙂
Thank you ever so much,
Maria Fernanda (Caracas – UCV).
Hello María Fernanda,
The problem I could find with merging both schemes is the cavity. When you optimize anything with a PCM cavity then you constrain the electron density (a main portion of it, anyway) to always be inside that cavity; If during the TS optimization some of it falls out then the program would stop due to an erroneous calculation of the solvation part, not necessarily that of the TS search.
If you were able to get to significantly similar results in both approaches I would advice you to cosider that is not always the case.
Of course the more complete method would be to use the solvent from the beginning but if for some reason PCM part crashes then I would settle for the second approach.
I hope this helps. Have a nice day!
Dear Dr. Joaquin,
Thank-you so much for your blog, its helped me a lot throughout my Ph.D. and the supervision of other students.
Recently I’ve been attempting to do an IRC calculation with no success. My system consists of a protein active site and as such, i have used modredundant to fix some coordinates. However, I appear to be encountering some errors in my calculations.
This is the error that i get:
” Charge and Multiplicity card seems defective:
Wanted an integer as input.”
Which doesn’t make sense to me as my charge and multiplicity are inputted correctly, therefore, I’m wondering if this has something to do with using modredundant in an IRC calculation.
What are your thoughts on this?
Many thanks,
Amy.
Dear Amy,
Thank you for your comments. In between coordinates for your QST3 or QST2 you have to add a line with a comment, typically indicating which structure belongs to products, reagents and the proposed TS. Maybe in your IRC you are missing the comment line or you lost a hydrogen atom somewhere along the way (that has happened to me before).
I hope this helps!
Hello Sir
In my reaction, according to the reactive sites available on the reactants, in principle, three kind of products can be possible. Experimentally, I am getting single product (regioselective product) and I know its structure, this means I know the structure of the product and reactants, so I tried QTS3 to know the structure transition state. I have optimized reactants and products both. Then using gauss view I have tried to generate new input file to know TS, but the final file is taking coordinates of only one compound either of any reactant or product.How to get input file using gauss view so that there will be no chance of any syntax error.
Thank you.
Copy your input here. You’re probably just missing a title or a blank line
%chk=file.chk
#N opt=(qst3,redundant) B3LYP/6-31++G(d,p) freq=noraman
Temperature=373.15 SCRF=(Solvent=Water)
ns-3
0 1
6 -3.322981000 0.446965000 0.017986000
6 -2.112976000 -0.291070000 0.002326000
6 -2.170461000 -1.685088000 0.063750000
6 -3.414765000 -2.312118000 0.145977000
6 -4.590842000 -1.563662000 0.162765000
6 -4.560985000 -0.166959000 0.097486000
6 -1.683668000 2.050918000 -0.125925000
6 -1.022446000 0.675156000 -0.082899000
1 -1.252614000 -2.252711000 0.040730000
1 -3.465238000 -3.393224000 0.193820000
1 -5.548510000 -2.068260000 0.225069000
1 -5.476881000 0.412313000 0.108632000
6 0.327868000 0.652244000 -0.115961000
6 1.225914000 -0.519607000 -0.089212000
6 2.706257000 -0.275226000 -0.017285000
6 3.552549000 -1.369112000 -0.252270000
6 3.277077000 0.966404000 0.295866000
6 4.931327000 -1.223816000 -0.190537000
6 4.660391000 1.108189000 0.368217000
6 5.489426000 0.017223000 0.120338000
7 -3.041638000 1.813107000 -0.057888000
1 -3.722782000 2.555660000 -0.068747000
8 0.802684000 -1.667804000 -0.142567000
8 -1.147127000 3.133711000 -0.200203000
1 3.099015000 -2.325186000 -0.481024000
1 5.574070000 -2.075483000 -0.382350000
1 6.566465000 0.131696000 0.170929000
1 5.089216000 2.071760000 0.618201000
1 2.654106000 1.826693000 0.503460000
1 0.780044000 1.635085000 -0.185075000
ns-bae
0 1
6 4.389021000 -1.398325000 0.042167000
6 4.833936000 -0.080302000 0.119150000
6 3.905648000 0.953208000 0.031057000
6 2.548913000 0.691267000 -0.138202000
6 2.099957000 -0.632942000 -0.196810000
6 3.036117000 -1.673455000 -0.108462000
7 0.752074000 -1.005751000 -0.396604000
6 -0.435297000 -0.392562000 -0.088647000
6 -1.615495000 -1.026922000 -0.317082000
6 -0.357092000 0.934293000 0.663154000
8 -0.123525000 0.931967000 1.837135000
8 -0.429993000 2.082848000 -0.031074000
6 -0.913076000 2.104789000 -1.386760000
6 -2.904079000 -0.438977000 0.007772000
8 -3.102736000 0.704588000 0.374539000
8 -3.905250000 -1.337707000 -0.164396000
6 -5.225521000 -0.855725000 0.130923000
1 5.094318000 -2.218730000 0.109507000
1 5.887729000 0.137174000 0.244635000
1 4.235444000 1.984469000 0.083822000
1 1.861952000 1.520305000 -0.222648000
1 2.694897000 -2.703379000 -0.146192000
1 0.643433000 -1.963653000 -0.700052000
1 -1.619783000 -2.033043000 -0.721243000
1 -1.987339000 1.927944000 -1.390433000
1 -0.399613000 1.369405000 -2.010282000
1 -0.693867000 3.106125000 -1.751584000
1 -5.888406000 -1.701552000 -0.038975000
1 -5.489644000 -0.025114000 -0.526399000
1 -5.289795000 -0.519338000 1.166877000
ns-amino
0 1
6 2.903982000 -1.128216000 0.574678000
6 4.016603000 -1.285239000 -0.264360000
6 5.312170000 -1.124480000 0.203952000
6 5.480614000 -0.790294000 1.551666000
6 4.384743000 -0.626410000 2.395733000
6 3.083377000 -0.798095000 1.905513000
6 1.651071000 -1.410160000 -0.228145000
6 2.206755000 -1.625754000 -1.661513000
1 6.167544000 -1.246347000 -0.450561000
1 6.483492000 -0.656757000 1.941614000
1 4.538858000 -0.366414000 3.436395000
1 2.227981000 -0.665470000 2.558849000
6 0.514012000 -0.431383000 -0.144485000
6 -0.815269000 -0.772836000 0.061055000
6 0.582921000 0.990030000 -0.171807000
6 -0.701056000 1.479578000 0.027422000
1 1.261014000 -2.392339000 0.066828000
8 1.573169000 -1.826104000 -2.672364000
7 3.582257000 -1.614318000 -1.552134000
1 4.172073000 -1.655025000 -2.369160000
6 -2.991462000 0.453363000 0.159712000
6 -3.666208000 0.056881000 -0.993499000
6 -3.696070000 0.880971000 1.282206000
6 -5.087176000 0.906798000 1.247772000
6 -5.769681000 0.505708000 0.100913000
6 -5.057932000 0.081951000 -1.019065000
7 -1.550672000 0.398098000 0.182266000
6 -1.189200000 2.873449000 0.050268000
6 -0.876608000 5.027476000 -0.846907000
8 -2.049925000 3.300306000 0.784710000
8 -0.554858000 3.626476000 -0.869532000
1 -0.284223000 5.472630000 -1.643013000
1 -1.941399000 5.178628000 -1.028753000
1 -0.614089000 5.463070000 0.118747000
1 -5.637108000 1.244846000 2.118340000
1 -6.853316000 0.526014000 0.079442000
1 -5.583189000 -0.228113000 -1.914927000
1 -3.155376000 1.209536000 2.159143000
6 -1.417638000 -2.116295000 0.145536000
6 -2.218985000 -2.494807000 1.232865000
6 -1.168042000 -3.052411000 -0.870223000
6 -2.759782000 -3.774156000 1.300784000
6 -1.707243000 -4.334571000 -0.793330000
6 -2.505642000 -4.698322000 0.288313000
6 1.823848000 1.765612000 -0.374360000
6 3.060323000 3.673231000 0.238714000
8 1.894060000 2.852479000 0.420977000
8 2.700002000 1.448172000 -1.144982000
1 3.108991000 4.046387000 -0.785684000
1 2.948962000 4.495395000 0.942396000
1 3.965104000 3.103171000 0.453602000
1 -0.557802000 -2.764198000 -1.719486000
1 -1.507074000 -5.046391000 -1.586295000
1 -2.927505000 -5.695586000 0.343740000
1 -3.375948000 -4.052030000 2.148549000
1 -2.410763000 -1.787081000 2.030167000
1 -3.101470000 -0.270725000 -1.857778000
Sir the input file.
The atom numbering is all screwed up. First eight atoms in the first coordinate set are C but in the second set only the first are C and then your seventh atom is N. The atomic numbering scheme should be kept constant throughout all three structures or there is no way for the program to follow each atom.
I suggest you define one with a visualizer like GaussView and then modify it appropriately to get the other two; that way the numbering remains the same.
I hope this helps
sir I followed this video to get input file, but in my case the input file has coordinates of only one single compound and also TS(QTS2) button was inactive. I run it multiple times but no improvement.
Hi Sir
I used the qst2 method to find the TS
After obtaining the TS, I did a freq calculation to check for Imaginary frequency and there was one at -283.17
To further confirm this ts, I did an IRC calculation and this is where it all went wrong
While visualing the IRC path, the reactant, product and TS had different energies that the ones I had already optimised earlier.
Can you please help?
Been stuck for more than 4 days…