
The energy of your calculated transition state (TS) is lower than that of the reagents. That’s gotta be an error right? Well, maybe not.
Typically, in classical transition state theory, we associate the reaction barrier to the energy difference between the reaction complex and the TS, in other words, we associate the reaction barrier to the relative energy of the TS. However, this isn’t always the case, since the TS isn’t always located at the barrier, which simply may not exist or may be a submerged one, i.e. the TS relative energy is negative with respect to the reaction complex. This leads to negative activation energies, but one must bear in mind that the activation energy is not equal to the relative energy of the TS but rather to the slope of the Arrhenius plot, which in turn comes from the Arrhenius equation given below.
k = Aexp(Ea/RT) or in logarithmic form Lnk = LnA + (Ea/RT) The Arrhenius plot is then the plot of Lnk vs T-1, with slope Ea
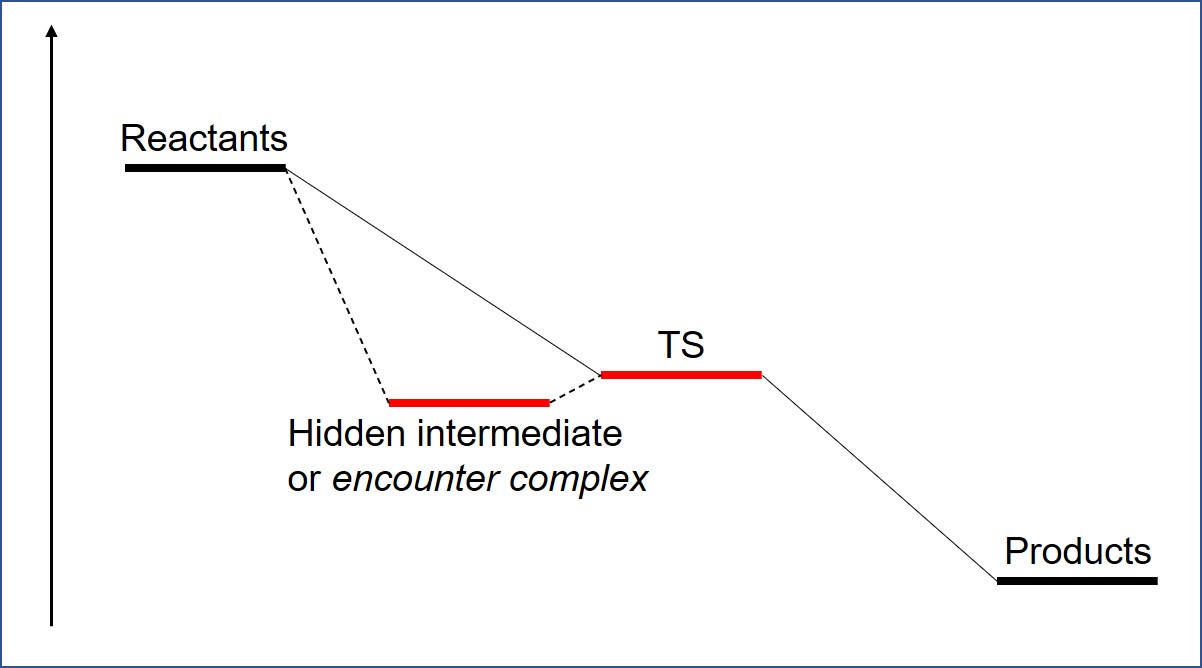
Caution is advised since the apparent presence of such a barrier may be due to a computational artifact rather than to the real kinetics taking place, that’s why an IRC calculation must follow a TS optimization in order to verify the truthfulness of the TS; keep in mind that in classical transition state theory, we’re ‘slicing‘ a multidimensional map along a carefully chosen reaction coordinate but this choice might not entirely be the right one, or even an existing one for that matter. I also recommend to change the level of theory, reconsider the reaction complex structure (because a hidden intermediate or complex may be lurking between reactants and TS, see figure 1) and fully verifying the thermochemistry of all components involved before asserting that any given reaction under study has one of these atypical barriers.

This is a very interesting and exceptional phenomena. Can you please send me a some relevant reference?