
As is the case of proteins, the functioning of DNA is highly dependent on its 3D structure and not just only on its sequence but the difference is that protein tertiary structure has an enormous variety whereas DNA is (almost) always a double helix with little variations. The canonical base pairs AT, CG stabilize the famous double helix but the same cannot be guaranteed when non-canonical -unnatural- base pairs (UBPs) are introduced.
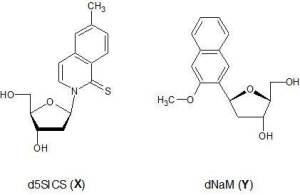
When I first took a look at Romesberg’s UBPS, d5SICS and dNaM (throughout the study referred to as X and Y see Fig.1) it was evident that they could not form hydrogen bonds, in the end they’re substituted naphtalenes with no discernible ways of creating a synton like their natural counterparts. That’s when I called Dr. Rodrigo Galindo at Utah University who is one of the developers of the AMBER code and who is very knowledgeable on matters of DNA structure and dynamics; he immediately got on board and soon enough we were launching molecular dynamics simulations and quantum mechanical calculations. That was more than two years ago.
Our latest paper in Phys.Chem.Chem.Phys. deals with the dynamical and structural stability of a DNA strand in which Romesberg’s UBPs are introduced sequentially one pair at a time into Dickerson’s dodecamer (a palindromic sequence) from the Protein Data Bank. Therein d5SICS-dNaM pair were inserted right in the middle forming a trisdecamer; as expected, +10 microseconds molecular dynamics simulations exhibited the same stability as the control dodecamer (Fig.2 left). We didn’t need to go far enough into the substitutions to get the double helix to go awry within a couple of microseconds: Three non-consecutive inclusions of UBPs were enough to get a less regular structure (Fig. 2 right); with five, a globular structure was obtained for which is not possible to get a proper average of the most populated structures.


X and Y don’t form hydrogen bonds so the pairing is pretty much forced by the scaffold of the rest of the DNA’s double helix. There are some controversies as to how X and Y fit together, whether they overlap or just wedge between each other and according to our results, the pairing suggests that a C1-C1′ distance of 11 Å is most stable consistent with the wedging conformation. Still much work is needed to understand the pairing between X and Y and even more so to get a pair of useful UBPs. More papers on this topic in the near future.
