
Calculating the pKa value for a Brønsted acid is very hard, like really hard. A full thermodynamic cycle (fig. 1) needs to be calculated along with the high-accuracy solvation free energy for each of the species under consideration, not to mention the use of expensive methods which will be reviewed here in another post in two weeks time.
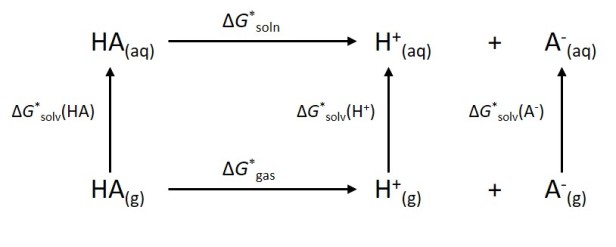
- Fig 1. Thermodynamic Cycle for the pKa calculation of any given Bronsted acid, HA
Finding descriptors that help us circumvent the need for such sophisticated calculations can help great deal in estimating the pKa value of any given acid. We’ve been interested in the reactivity of σ-hole bearing groups in the past and just like Halogen, Tetrel, Pnicogen and Chalcogen bonds, Hydrogen bonds are highly directional and their strength depends on the polarization of the O-H bond. Therefore, we suggested the use of the maximum surface electrostatic potential (VS,max) on the acid hydrogen atom of carboxylic acids as a descriptor for the strength of their interaction with water, the first step in the deprotonation process.
We selected six basis sets; five density functionals; the MP2 method for a total of thirty-six levels of theory to optimize and calculate VS,max on thirty carboxylic acids for a grand total of 1,080 wavefunctions, which were later passed onto MultiWFN (all calculations were taken with PCM = water). Correlation with the experimental pKa values showed a great correlation across the levels of theory (R2 > 0.9), except for B3LYP. Still, the best correlations were obtained with LC-wPBE/cc-pVDZ and wB97XD/cc-pVDZ. From this latter level of theory the linear correlation yielded the following equation:
pKa = -0.2185(VS,max) + 16.1879
Differences in pKa turned out to be less than 0.5 units, which is remarkable for such a straightforward method; bear in mind that calculation of full thermodynamic cycles above chemical accuracy (1.0 kcal/mol) yields pKa differences above 1.0 units.
We then took this equation for a test with 10 different carboxylic acids and the prediction had a correlation of 98% (fig. 2)
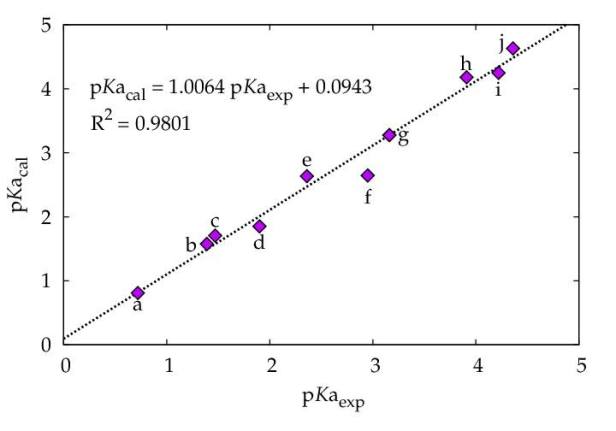
- fig 2. calculated v experimental pKa values for a test set of 10 carboxylic acids from equation above
I think this method can really catch on for a quick way to predict the pKa values of any carboxylic acid imaginable. We’re now working on the model extension to other groups (i.e. Bronsted bases) and putting together a black-box workflow so as to make it even more accessible and straightforward to use.
We’ve recently published this work in the journal Molecules, an open access publication. Thanks to Prof. Steve Scheiner for inviting us to participate in the special issue devoted to tetrel bonding. Thanks to Guillermo Caballero for the inception of this project and to Dr. Jacinto Sandoval for taking the time from his research in photosynthesis to work on this pet project of ours and of course the rest of the students (Gustavo Mondragón, Marco Diaz, Raúl Torres) whose hard work produced this work.

Dear Joaquin,
Here it is my experience by the topic stated by you, hope to like it – Ionization and Conformational Equilibria of Citric Acid: Delocalized Proton Binding in Solution,
The Journal of Physical Chemistry A 121(31),
best regards
Mira
Dear Mira, I liked it very much! I will keep it in mind for further work. Happy new year! Stay in touch
hello, i’m student from algeria, please help me, wh
This is an interesting work. Please check out our recent work also on pKa, published on J. Mol. Model.
Correlation between molecular acidity (pKa) and vibrational spectroscopy
https://link.springer.com/article/10.1007/s00894-019-3928-4
Did you cite our work? LOL just kidding! Looks very interesting and thorough, thanks for bringing it to our attention. I’ll read it in depth tomorrow and will come back to you with questions. Have a nice day!
Thanks Dr. Barroso.