
Happy new year to all my readers!
Having a new paper published is always a matter of happiness for this computational chemist but this time I’m excedingly excited about anouncing the publishing of a paper in the Journal of Chemical Theory and Computation, which is my highest ranked publication so far! It also establishes the consolidation of our research group at CCIQS as a solid and competitive group within the field of theoretical and computational chemistry. The title of our paper is “In Silico design of monomolecular drug carriers for the tyrosine kinase inhibitor drug Imatinib based on calix- and thiacalix[n]arene host molecules. A DFT and Molecular Dynamics study“.
In this article we aimed towards finding a suitable (thia-) calix[n]arene based drug delivery agent for the drug Imatinib (Gleevec by Novartis), which is a broadly used powerful Tyrosine Kinase III inhibitor used in the treatment of Chronic Myeloid Leukaemia and, to a lesser extent, Gastrointestinal Stromal Tumors; although Imatinib (IMB) exhibits a bioavailability close to 90% most of it is excreted, becomes bound to serum proteins or gets accumulated in other tissues such as the heart causing several undesired side effects which ultimately limit its use. By using a molecular capsule we can increase the molecular weight of the drug thus increasing its retention, and at the same time we can prevent Imatinib to bind, in its active form, to undesired proteins.
We suggested 36 different calix and thia-calix[n]arenes (CX) as possible candidates; IMB-CX complexes were manually docked and then optimized at the B97D/6-31G(d,p) level of theory; Stephan Grimme’s B97D functional was selected for its inclusion of dispersion terms, so important in describing π-π interactions. Intermolecular interaction energies were calculated under the Natural Bond Order approximation; a stable complex was needed but a too stable complex would never deliver its drug payload! This brings us to the next part of the study. A monomolecular drug delivery agent must be able to form a stable complex with the drug but it must also be able to release it. Molecular Dynamics simulations (+100 ns) and umbrella sampling methods were used to analyse the release of the drug into the aqueous media.
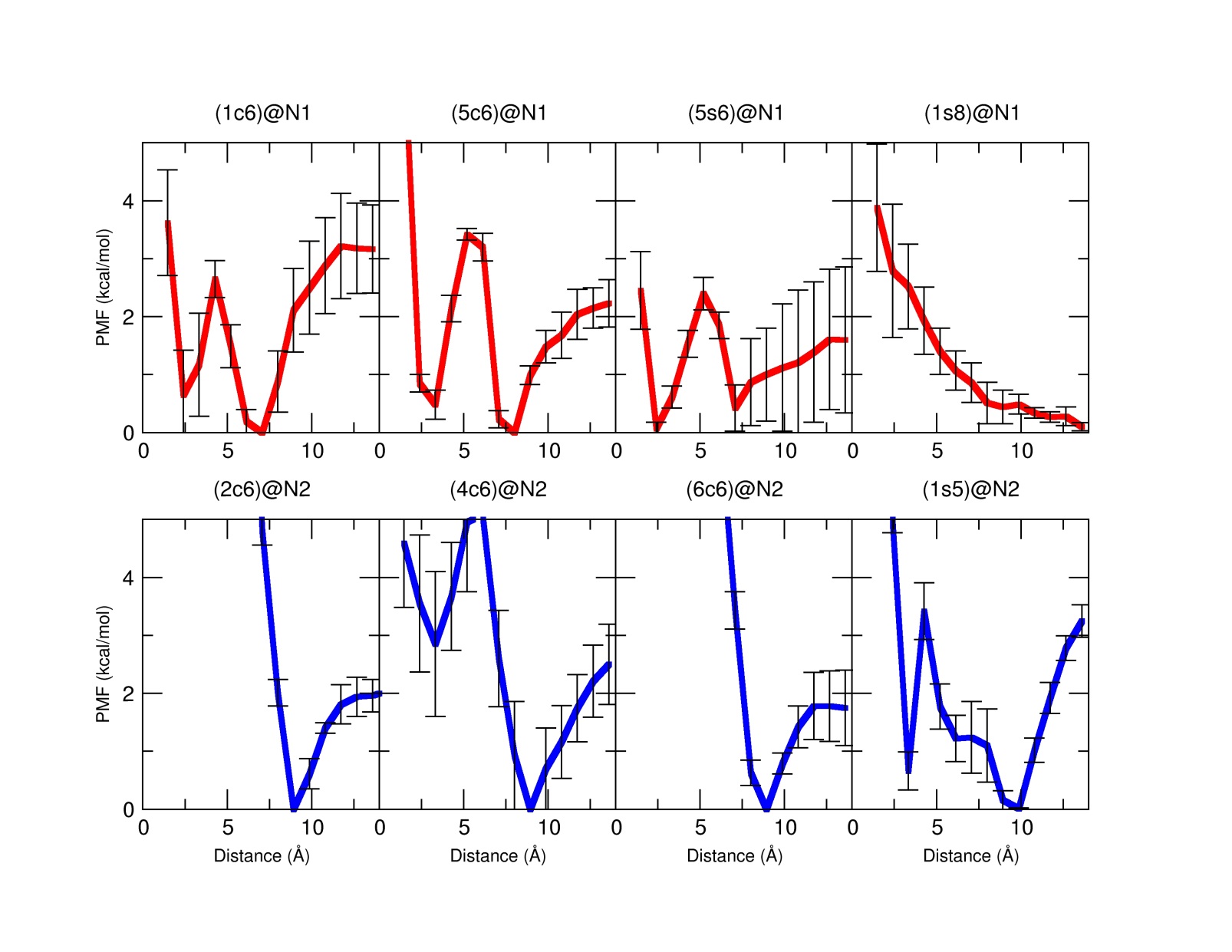
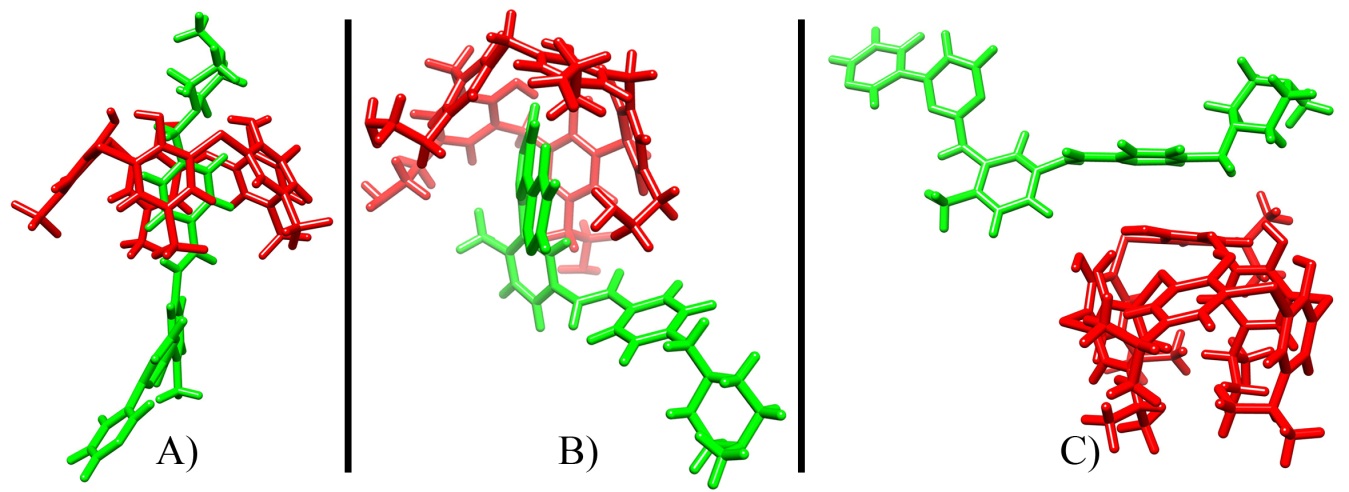
Potential Mean Force profiles for the four most stable complexes for position N1 and N2 from the QM simulations are shown below (Red, complexes in the N1 position, blue, N2 position). These plots, derived from the MD simulations give us an idea of the final destination of the drug respect of the calixarene carrier. In the next image, the three preferred structures (rotaxane-like; inside; released) for the final outcome of the delivery process are shown. The stability of the complexes was also assessed by calculating the values of ΔG binding through the use of the Poisson equations.
Thanks to my co-authors Maria Eugenia Sandoval-Salinas and Dr. Rodrigo Galindo-Murillo for their enormous contributions to this work; without their hard work and commitment to the project this paper wouldn’t have been possible.

congratulation sir….
Thank you very much!
I liked your figures. Which software did you use for making the figures ?
GNUplot for the PMF analysis, gaussview for the molecular structures (joined simply in mspowerpoint)
Thanks for your message!
Que buen articulo…
Muchas gracias!! saludos
I congratulate you for this marvelous work you did and wish you would benefit more than the paper. I have a question, it could seem so simple to be asked., but I need your help. A IRC job an doing now on crude oil molecules in order to understand more the heteroatoms removing, does not complete normally and gives in the output file this:Pt 27 Step number 20 out of a maximum of 20. The calculation is done with Guaussian09 and I specified 25 as computed points and 10 as stepsize. Thanks a lot.
Congratulations on your highly relevant and insightful publication.
Thanks, Greg!
Have a nice day
Hello Mr BARROSO. I need your help and really. I use in my works WS2 modeled as WS3H3+, but I want to promote it with Ni and Co. None of all the papers I read gave me the structure to model it in Gaussview. I would you to direct me in realising this step whitch is the first and important one of what I planed to do. Thanks a lot
Finally i quit my regular job, now i earn a lot
of money online you should try too, just type in google – blackhand roulette system
I read a lot of interesting articles here. Probably you spend a lot
of time writing, i know how to save you
a lot of time, there is an online tool that creates readable, SEO friendly articles in minutes,
just type in google – laranitas free content source