
Editing large molecules on a seemingly simple visualizer as GaussView can be a bit daunting. I’m working on a follow up of that project we recently published in JACS but now we require to attach two macrocycles to the organometallic moiety; the only caveat is that this time we don’t have any crystallographic data with which to start. Generating a 3D model of this structure is already hard enough and even when you managed to do it there are many degrees of freedom that in some cases can lead to unrealistic geometries after optimization.
I recently came across a simple way to edit a large complicated molecule by optimizing the fragments separately and then joining them in a new molecule by using the clipboard. This rather simple method, that I for one had never exploited has just saved me a few good hours.
Copy a molecule (CTRL+C) and it will go to the clipboard as a molecular fragment for which you can define a new hot atom and thus bind it to the other fragment as you would with the regular builder. I strongly suggest to use a “New Molecule Group” instead of editing over an existing molecule. Also, if you are using the “paste” button, observe that it has three different options; you may want to use the last one “append to existing molecule” or you will have your fragments in different windows.
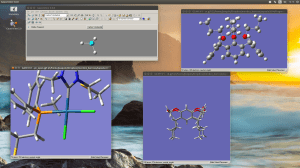
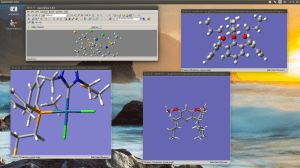
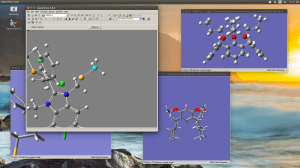
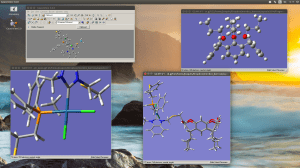
And remember, dihedral angles are your best friends.
