
Some atomic properties such as an atomic charge are isotropic, but every now and then some derivations of them become anisotropic, for example the plotting of the Molecular Electrostatic Potential (MEP) on the electron density surface can exhibit some anisotropic behavior; quantifying it can be a bit challenging.
It is well known that halogen atoms such as Chlorine can form so-called halogen-bonds of the type R-Cl-R in crystals with a near perfect 180° angle. This finding has lead to the discovery of σ-holes in halogens. σ-Holes are electrophilic portions of the anisotropic electrostatic potential in an otherwise nucleophilic atom. Recently, Guillermo “Memo” Caballero and I calculated the MEP for a series of trichloromethyl-containing compounds at the MP2/cc-pVQZ level of theory and the mapping shows evidence of such σ-holes as seen in Figure1. Those small blue portions on an otherwise red atom indicate that some electron density is missing in that position, which by the way is located at 180° away from the carbon atom.


But having the picture is not enough. We want to quantify just how strong are those σ-holes to effectively attract a nucleophile and perhaps perform some chemistry on the C-Cl bond. That’s when we resorted to MultiWFN, a Multifunctional Wavefunction Analyzer developed by Tian Lu (卢天) at the Beijing Kein Research Center for Natural Sciences. You can check the project leader list of publications here. Among many other capabilities, MultiWFN is able to print details about properties along a surface.
In order to work with MultiWFN you need to generate a *.wfn file, if you have a previous Gaussian calculation for which you want to analyze their surface you can run a guess=only calculation in order to extract the wavefunction from the checkpoint file. Here is a dummy of the input for such calculation
%chk=oldfile.chk # output=wfn geom=check guess=(only,read) density=current --blank-- Title Card --blank-- 0 1 --blank-- filename.wfn --blank--
In our case, having a post-Hartree-Fock calculation, the use of density=current is mandatory to get the MP2 density matrix and not just the HF one. Running this calculation will generate the file filename.wfn which is now used with MultiWFN. When starting MultiWFN you get to see a terminal window like the one below in which you are asked to input the path of your wfn file:

After loading it you will get the following window with the various options available. Type 12// (these two slashes are mandatory) to get the quantitative analysis of molecular surface option.
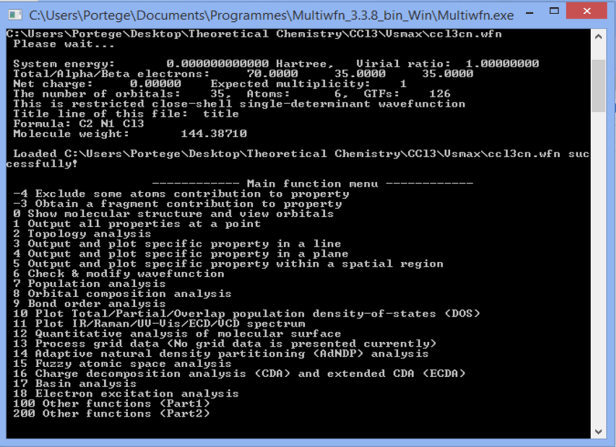
Then you will be asked to define some elements of that surface (we used the default options 0)
The following screenshot shows the results section in which several maxima and minima of electrostatic potential were found (7 and 11 in our case); a star is placed on the side of the global maximum. The value of the MEP at those points is given in Hartrees, eV and kcal/mol which I personally hate because there isn’t such a thing as a mole of ‘potentials’ (same argument as giving an orbital’s energy in kcal/mol, moles of what? orbitals? Personally, I don’t like it even if its valid).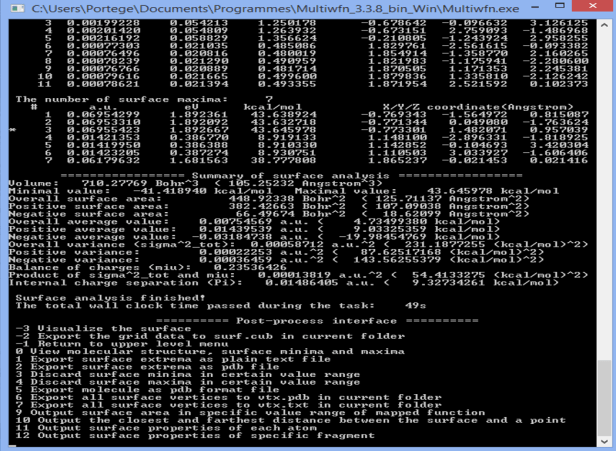
Their visualizer is activated through the option 0 and although it is far from pretty it is quite good enough to find the numbers corresponding to maxima and minima of the MEP on the isodensity surface. If we look for the maxima then we find for our example (CHCl3) that a maximum is located in front of each Cl atom in a straight line from the C atom. Now we get to put a number on the mapped isosurface provided by Gaussian or even import the file into Chimera.


We are still working our way around MultiWFN, I hope we can find the batch option, it would be most useful. In the mean time, Guillermo and I will continue to search for σ-holes in chlorinated reagents. Thanks to Guillermo for his ongoing work in this and other topics within the realm of organic reactivity.
Have you any suggestions or ideas to work with MultiWFN? Please share them!

First of all I want to thanks , many times I have looked in your blog searching for help.
When I deal with multiwfn on linux I use bash scripts and a scheduler (such torque) to have a batch. The options can be written in a file and passed to the program, while the outputs that can’t be automatically exported could be stored in a txt file.
I’m not sure if it is helpful.
Marco
Hi Marco,
Thank you for your post and for me it is very useful, I want to know more about scripting Multiwfn into bash. Specially, I want to script the following steps: Multiwfn file.fchk <<< 8 1 1 and save it to a file.
Thank you so much in advance for any reply.
David
You can add below lines into your script file:
Multiwfn file.fchk > out.txt << EOF
8
1
1
EOF
Another way to realize your purpose: echo -e “8\n1\n1\n” | Multiwfn file.fchk > out.txt
where \n corresponds to pressing ENTER button
This only works for Linux or MacOS system.
Dear Tian Lu
Thank you very much for your input!
Hi,
I’m developer of Multiwfn program, thank you very much for introducing Multiwfn to more people. I have several comments about this post:
1 In fact “//” is never needed, you can simply input “12”. The // in the manual is only used to denote that the texts after // are comment.
2 The kcal/mol is frequently employed in literature as unit of MEP, see e.g. J Mol Model (2008) 14:659–665, because its magnitude is very suitable to represent MEP. It is simply an energy unit, and you don’t need to carefully concern about its physical meaning.
3 When you visualize surface extrema in the GUI of Multiwfn, you can enlarge “Ratio of atomic size” to 4.0, then the atomic spheres will correspond to atomic vdW spheres; in this case it is easier to examine the position of surface extrema.
4 It is very easy to run Multiwfn in batch mode by redirection and shell script, please check Section 5.2 and 5.3 of Multiwfn manual.
Tian
Dear Joaquín,
I have implemented a kind of plots using the intrinsic polarizability, that is, a size-independent polarizability based on atomic partitionings like Hirshfeld-baded and QTAIM partitionings (if you are interested, see the article http://onlinelibrary.wiley.com/doi/10.1002/jcc.24003/abstract). You may be interested in studying how the inner shells of the atoms are affected by the polarization to compare with your MEP plots. A collaboration is also possible. I only need either your geometries and calculation level or your fchk files to obtain my plots in cube format.
By the way, thank you very much Tian. By means of Multiwfn I have obtained the QTAIM intrinsic polarizability plots easily.
NICO
I would like 2016 to be the year when people remembered that science is a method of investigation,and NOT a belief system
How to generate WFN file?
Dear Abisha.W Wilson,
The way of generating .wfn file by several popular quantum chemistry codes has been detailedly described at the beginning of Chapter 4 of Multiwfn manual, which is available at http://sobereva.com/multiwfn, please have a check.