
We’ve expanded the scope of our research interests from quantum mechanical calculations to docking and MedChem for over a year now; it has been a very interesting ride and a very rich avenue of research to explore. Durbis Castillo has led -out of his own initiative- this project and today he presents us with a guest post on the nuances of his project. Bear in mind that the detail of the calculations and a small -very targeted- tutorial on MAESTRO will be provided later in further posts and that making all this decisions required a long process of trial and error, we can only thank Dr. Antonio Romo for his help in minimizing the time this process took.
HIV is a tricky virus, and even though many of the steps included in its lifecycle are druggable, the chemical machinery making it work has been quite elusive since research groups started studying it. Highly Active Antiretroviral Therapy (HAART) works thanks to the combination of several drugs targeting different proteins such as the HIV protease or reverse transcriptase.
In 1998 the elucidation of the gp120 envelope glycoprotein crystal structure introduced a new step in the drug discovery race: HIV entry. Since drugs targeting gp120 have not been widely explored or developed, we decided to use common methodologies like docking (rigid and fit-induced) and ADME predictions to address the following question: How can we easily discover a molecule that inhibits gp120 binding to the lymphocyte CD4 receptor without having to synthesize it first? The answer was to perform a virtual screening with a bottleneck methodology based on docking calculations.
Docking methodologies are often looked as insufficient, careless or even unscientific, since the algorithms they are founded upon are not as accurate or descriptive as the ones that support DFT or ab initio calculations, for example. But there is a huge advantage to simpler operations: less computational resources are required. Then, following Russia’s example when making tanks during the WWII, why not make thousands or millions of docking calculations to quickly explore an entire chemical space and find which molecules are more likely to bind the protein?
And this is exactly what we did. We built a piperazine-based dataset of 16.3 million compounds, all of them including fragments that are reported in the medicinal chemistry literature, thus having two main characteristics, synthetic accessibility and pharmacological activity. These 16.3 million compounds were thoroughly filtered through several docking steps, each one of them being more accurate and comprehensive than the previous one, abruptly eliminating poorly fitted molecules, leaving us with a total of 275 candidates that were redocked in a different crystal structure and a different program (consensus docking).
After analyzing the ADME properties of the candidates, with descriptors such as human oral absorption and possible metabolic reactions, as well as the Induced-Fit Docking score of these molecules, ten ligands were selected as the best ones inside the analyzed chemical space. You can see ligand 255 (figure 1) as an example of the molecules that obtained the best scores throughout the docking steps.
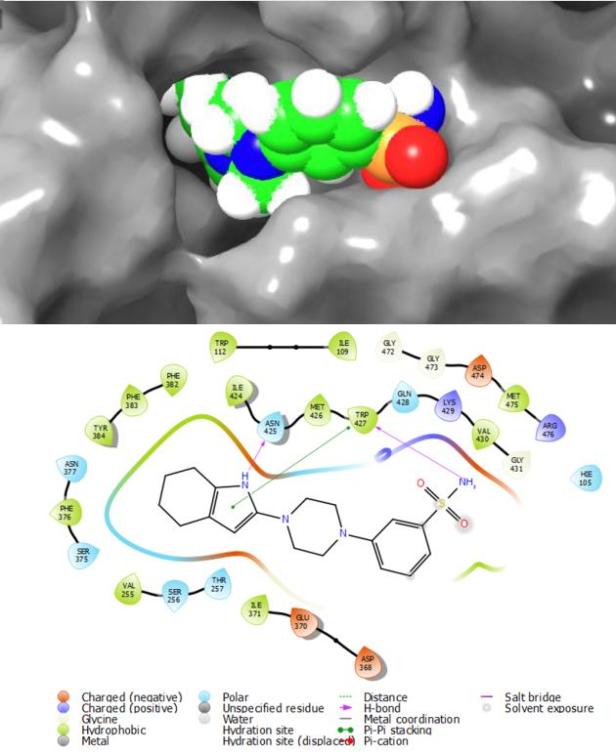
Many of the colleague researchers related to this kind of topics asked “Why didn’t you download a set of molecules from Zinc or Maybridge?” And the answer to this question includes three aspects: first we wanted to test a combinatorial approach to drug design, second, we wanted to test whether including a piperazine as the core of the set of molecules would immediately grant them activity and high potency, and finally, a built database will always confer a higher degree of novelty to the possible hits when compared to commercially available compounds whose synthesis has already been developed. However, this last point needs to be addressed by an organic chemist since none of the molecules from our database have ever been synthesized (any takers?).
Right now, we are trying to explore further through molecular dynamics simulations using Desmond and Amber. Other future goals for this project include screening large databases of commercial and novel compounds with gp120 and other proteins involved in the HIV lifecycle. Also, we remain open to collaborate with anyone interested in taking the challenge to synthesize our molecules, as well as performing the biochemical assays to get an idea of their activity.
More details on MD simulations and the path of our first virtual hits to follow. Anyone interested in reading my thesis work can contact me through my linkedin profile at https://www.linkedin.com/in/durbisjaviercp/. An article is under preparation and will soon be submitted, stay tuned!

You need to hire my HIGH SCHOOL students…we teach both a full semester course in computational quantum chemistry AND computational medicinal chemistry, including lots of docking!
Sounds like a good idea. Thanks for reading!
Please contact me for more details; we should be able to come up with a plan for them. Thanks for the heads up
Dear Dr. Joaquin,
How can i contact your personally?
On Thu, Jan 18, 2018 at 9:16 PM, Dr. Joaquin Barroso’s Blog wrote:
> joaquinbarroso posted: “We’ve expanded the scope of our research interests > from quantum mechanical calculations to docking and MedChem for over a year > now; it has been a very interesting ride and a very rich avenue of research > to explore. Durbis Castillo has led -out of his own ” >
send me an email to jbarroso -at- unam-dot-mx