Calculating reaction constants, in the correct physical way, requires accurate thermodynamic calculations involving solvation/desolvation energies at high levels of theory, because free energy deviations lower than 1 kcal/mol lead to reaction constant values off by an order of magnitude.
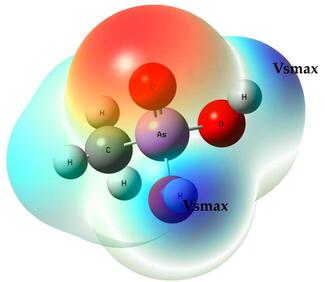
We’ve been capable to calculate pka for carboxylic acids and pkb values for amino groups accurately by using an electrostatic approach based on calculating the electrostatic potential over an isosurface of the atoms of interest (H and N, respectively). The maximum value of electrostatic potential on the atom’s isosurface is labeled as Vs,max (or Vs,min for negative values as the ones located on basic atoms), and Vs,max correlates
Designing strategies for capturing pollutants, such as arsenic based acids, greatly benefits from knowing in advance their physicochemical properties like their pka. Therefore, predicting these pka values accurately but rapidly poses an advantage for environmental scientists and engineers.
In this study, we used four different models to calculate pKa values for a family of thirty five arsonic acids (RAsO3H2). Three of these methods were based on DFT (including our previously favored Vs,max) and a fourth one based on Machine Learning (Support Vector Machines).
Vs,max: We calculated the electrostatic potential over the isosurface of the acidic hydrogen atoms and these values were correlated with experimental pka’s and we got a MAE of 0.25 units, not too bad but definitely not the best correlations were obtained as R2 values did not exceed 0.60.
(Scaled) Solvent Accessible Surface (sSAS): Solvation energies are required to calculate the thermodynamic cycle for the deprotonation reaction, however, they are quite sensitive to the continuum solvation model used. Optimizing (scaling) the solvent accessible surface for the SMD model improves calculation of pka’s but still we weren’t able to accurately reproduce the experimental values (R2 = 0.45)
Atomic Charge-Based Model: A benchmark for carboxylic acids was reported by Monard calculating NPA charges with the CPCM solvation model, and we adapted it to our arsonic acids by averaging the atomic charges of all three oxygen atoms at their anionic state. This turned out to be the winner! we got R2 = 0.9654 and 0.9827 for the monoanionic and bisanionic states, respectively.
Machine Learning (SVM): Finally, we took the ML approach using Alvadesc with a 4,000 descriptors categorized in 22 classes. Upon optimizing the various combinations of descriptors, the highest R2 value obtained was 0.67 with a value of MAE = 0.48.
This goes to show that we should never stick to one model when calculating physicochemical properties. In full disclosure, this study began as a sort of bet between our Vs,max approach and the ML methods. Maybe ML would’ve worked better if we had a larger set (35 molecules seem to be very few), and maybe Vs,max calculations fail due to some unaccounted interaction with arsenic’s orbitals.
Read the full paper free of charge in ACS Omega (Comparative Analysis of pKa Predictions for Arsonic Acids Using Density Functional Theory-Based and Machine Learning Approaches DOI:10.1021/acsomega.4c10413). Thanks to my good friend Dr. Miroslava Nedyalkova from the University of Fribourg, Switzerland, for instigating this collaboration.
