
Literature in synthetic chemistry is full of reactions that do occur but very little or no attention is payed to those that do not proceed. The question here is what can we learn from reactions that are not taking place even when our chemical intuition tells us they’re feasible? Is there valuable knowledge that can be acquired by studying the ‘anti-driving force’ that inhibits a reaction? This is the focus of a new manuscript recently published by our research group in Tetrahedron (DOI: 10.1016/j.tet.2016.05.058) which was the basis of Guillermo Caballero’s BSc thesis.

It is well known in organic chemistry that if a molecular structure has the possibility to be aromatic it can somehow undergo an aromatization process to achieve this more stable state. During some experimental efforts Guillermo Caballero found two compounds that could be easily regarded as non-aromatic tautomers of a substituted pyridine but which were not transformed into the aromatic compound by any means explored; whether by treatment with strong bases, or through thermal or photochemical reaction conditions.
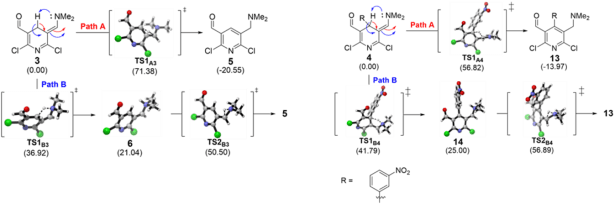
These results led us to investigate the causes that inhibits these aromatization reactions to occur and here is where computational chemistry took over. As a first approach we proposed two plausible reaction mechanisms for the aromatization process and evaluated them with DFT transition state calculations at the M05-2x/6-31+G(d,p)//B3LYP/6-31+G(d,p) levels of theory. The results showed that despite the aromatic tautomers are indeed more stable than their corresponding non-aromatic ones, a high activation free energy is needed to reach the transition states. Thus, the barrier heights are the first reason why aromatization is being inhibited; there just isn’t enough thermal energy in the environment for the transformation to occur.

But this is only the proximal cause, we went then to search for the distal causes (i.e. the reasons behind the high energy of the barriers). The second part of the work was then the calculation of the delocalization energies and frontier molecular orbitals for the non-aromatic tautomers at the HF/cc-pVQZ level of theory to get insights for the large barrier heights. The energies showed a strong electron delocalization of the nitrogen’s lone pair to the oxygen atom in the carbonyl group. Such delocalization promoted the formation of an electron corridor formed with frontier and close-to-frontier molecular orbitals, resembling an extended push-pull effect. The hydrogen atoms that could promote the aromatization process are shown to be chemically inaccessible.

Further calculations for a series of analogous compounds showed that the dimethyl amino moiety plays a crucial role avoiding the aromatization process to occur. When this group was changed for a nitro group, theoretical calculations yielded a decrease in the barrier high, enough for the reaction to proceed. Electronically, the bonding electron corridor is interrupted due to a pull-pull effect that was assessed through the delocalization energies.
The identity of the compounds under study was assessed through 1H, 13C-NMR and 2D NMR experiments HMBC, HMQC so we had to dive head long into experimental techniques to back our calculations.

hello doctor, I need help, I would like to know the external electric field value in gaussiane.la default value. is that the default value of the electric field if 00 ?. thank you in advance.
hola doctor, antes que nada felicitarle por su ardo trabajo en el área de investigación científica, y por otro lado me guata ria conocerle en persona no se si pueda darme esa oportunidad, mi nombre es Juan Nabor Balderas Gutiérrez estudiante actualmente del doctorado en ciencias ambientales en el instituto tecnológico de toluca, pero me llama la atención en área de simulación, ya que durante mis estudios de maestría presente un trabajo de simulación de estructuras moleculares de una molécula de pirrol y un átomo de hidrógeno, aunque esto no era mi trabajo de maestría, yo estoy mas enfocado en el área de sistemas generadores de plasma. sin mas por el momento me despido de usted de la manera mas atenta enviando de nuevo un cordial saludo.
El Miércoles, 15 de junio, 2016 9:38:39, Dr. Joaquin Barrosos Blog escribió:
#yiv7589309662 a:hover {color:red;}#yiv7589309662 a {text-decoration:none;color:#0088cc;}#yiv7589309662 a.yiv7589309662primaryactionlink:link, #yiv7589309662 a.yiv7589309662primaryactionlink:visited {background-color:#2585B2;color:#fff;}#yiv7589309662 a.yiv7589309662primaryactionlink:hover, #yiv7589309662 a.yiv7589309662primaryactionlink:active {background-color:#11729E;color:#fff;}#yiv7589309662 WordPress.com | joaquinbarroso posted: “Literature in synthetic chemistry is full of reactions that do occur but very little or no attention is payed to those that do not proceed. The question here is what can we learn from reactions that are not taking place even when our chemical intuition te” | |