
Calculation of interaction energies is one of those things people are more concerned with and is also something mostly done wrong. The so called ‘gold standard‘ according to Pavel Hobza for calculating supramolecular interaction energies is the CCSD(T)/CBS level of theory, which is highly impractical for most cases beyond 50 or so light atoms. Basis set extrapolation methods and inclusion of electronic correlation with MP2 methods yield excellent results but they are not nonetheless almost as time consuming as CC. DFT methods in general are terrible and still are the most widely used tools for electronic structure calculations due to their competitive computing times and the wide availability of schemes for including terms which help describe various kinds of interactions. The most important ingredients needed to get a decent to good interaction energies values calculated with DFT methods are correlation and dispersion. The first part can be recreated by a good correlation functional and the use of empirical dispersion takes care of the latter shortcoming, dramatically improving the results for interaction energies even for lousy functionals such as the infamous B3LYP. The results still wont be of benchmark quality but still the deviations from the gold standard will be shortened significantly, thus becoming more quantitatively reliable.
There is an online tool for calculating and adding the empirical dispersion from Grimme’s group to a calculation which originally lacked it. In the link below you can upload your calculation, select the basis set and functionals employed originally in it, the desired damping model and you get in return the corrected energy through a geometrical-Counterpoise correction and Grimme’s empirical dispersion function, D3, of which I have previously written here.
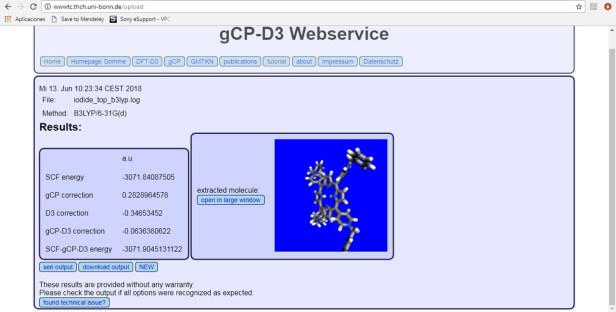
The gCP-D3 Webservice is located at: http://wwwtc.thch.uni-bonn.de/
The platform is entirely straightforward to use and it works with xyz, turbomole, orca and gaussian output files. The concept is very simple, a both gCP and D3 contributions are computed in the selected basis set and added to the uncorrected DFT (or HF) energy (eq. 1)
![]() (1)
(1)
If you’re trying to calculate interaction energies, remember to perform these corrections for every component in your supramolecular assembly (eq. 2)
 (2)
(2)
Here’s a screen capture of the outcome after uploading a G09 log file for the simplest of options B3LYP/6-31G(d), a decomposed energy is shown at the left while a 3D interactive Jmol rendering of your molecule is shown at the right. Also, various links to the literature explaining the details of these calculations are available in the top menu.
I’m currently writing a book chapter on methods for calculating ineraction energies so expect many more posts like this. A special mention to Dr. Jacinto Sandoval, who is working with us as a postdoc researcher, for bringing this platform to my attention, I was apparently living under a rock.

For users who want to maintain stability and use their phones very frequently can opt for these plans. fix phone shop