 Gaussian")
The canonical molecular orbital depiction of an electronic transition is often a messy business in terms of a ‘chemical‘ interpretation of ‘which electrons‘ go from ‘which occupied orbitals‘ to ‘which virtual orbitals‘.
Natural Transition Orbitals provide a more intuitive picture of the orbitals, whether mixed or not, involved in any hole-particle excitation. This transformation is particularly useful when working with the excited states of molecules with extensively delocalized chromophores or multiple chromophoric sites. The elegance of the NTO method relies on its simplicity: separate unitary transformations are performed on the occupied and on the virtual set of orbitals in order to get a localized picture of the transition density matrix.
[1] R. L. Martin, J. Chem. Phys., 2003, DOI:10.1063/1.1558471.
In Gaussian09:
After running a TD-DFT calculation with the keyword TD(Nstates=n) (where n = number of states to be requested) we need to take that result and launch a new calculation for the NTOs but lets take it one step at a time. As an example here’s phenylalanine which was already optimized to a minimum at the B3LYP/6-31G(d,p) level of theory. If we take that geometry and launch a new calculation with the TD(Nstates=40) in the route section we obtain the UV-Vis spectra and the output looks like this (only the first three states are shown):
Excitation energies and oscillator strengths: Excited State 1: Singlet-A 5.3875 eV 230.13 nm f=0.0015 <S**2>=0.000 42 -> 46 0.17123 42 -> 47 0.12277 43 -> 46 -0.40383 44 -> 45 0.50838 44 -> 47 0.11008 This state for optimization and/or second-order correction. Total Energy, E(TD-HF/TD-KS) = -554.614073682 Copying the excited state density for this state as the 1-particle RhoCI density. Excited State 2: Singlet-A 5.5137 eV 224.86 nm f=0.0138 <S**2>=0.000 41 -> 45 -0.20800 41 -> 47 0.24015 42 -> 45 0.32656 42 -> 46 0.10906 42 -> 47 -0.24401 43 -> 45 0.20598 43 -> 47 -0.14839 44 -> 45 -0.15344 44 -> 47 0.34182 Excited State 3: Singlet-A 5.9254 eV 209.24 nm f=0.0042 <S**2>=0.000 41 -> 45 0.11844 41 -> 47 -0.12539 42 -> 45 -0.10401 42 -> 47 0.16068 43 -> 45 -0.27532 43 -> 46 -0.11640 43 -> 47 0.16780 44 -> 45 -0.18555 44 -> 46 -0.29184 44 -> 47 0.43124
The oscillator strength is listed on each Excited State as “f” and it is a measure of the probability of that excitation to occur. If we look at the third one for this phenylalanine we see f=0.0042, a very low probability, but aside from that the following list shows what orbital transitions compose that excitation and with what energy, so the first line indicates a transition from orbital 41 (HOMO-3) to orbital 45 (LUMO); there are 10 such transitions composing that excitation, visualizing them all with canonical orbitals is not an intuitive picture, so lets try the NTO approach, we’re going to take excitation #10 for phenylalanine as an example just because it has a higher oscillation strength:
%chk=Excited State 10: Singlet-A 7.1048 eV 174.51 nm f=0.3651 <S**2>=0.000 41 -> 45 0.35347 41 -> 47 0.34685 42 -> 45 0.10215 42 -> 46 0.17248 42 -> 47 0.13523 43 -> 45 -0.26596 43 -> 47 -0.22995 44 -> 46 0.23277
Each set of NTOs for each transition must be calculated separately. First, copy you filename.chk file from the TD-DFT result to a new one and name it after the Nth state of interest as shown below (state 10 in this case). NOTE: In the route section, replace N with the number of the excitation of interest according to the results in filename.log. Run separately for each transition your interested in:
#chk=state10.chk #p B3LYP/6-31G(d,p) Geom=AllCheck Guess=(Read,Only) Density=(Check,Transition=N) Pop=(Minimal,NTO,SaveNTO) 0 1 --blank line--
By requesting SaveNTO, the canonical orbitals in the state10.chk file are replaced with the NTOs for the 10th excitation, this makes it easier to plot since most visualizers just plot whatever set of orbitals they read in the chk file but if they find the canonical MOs then one would need to do some re-processing of them. This is much more straightforward.
Now we format our chk files into fchk with the formchk utility:
formchk -3 filename.chk filename.fchk
formchk -3 state10.chk state10.fchk
If we open filename.fchk (the file where the original TD-DFT calculation is located) with GaussView we can plot all orbitals involved in excited state number ten, those would be seven orbitals from 41 (HOMO-3) to 47 (LUMO+2) as shown in figure 1.
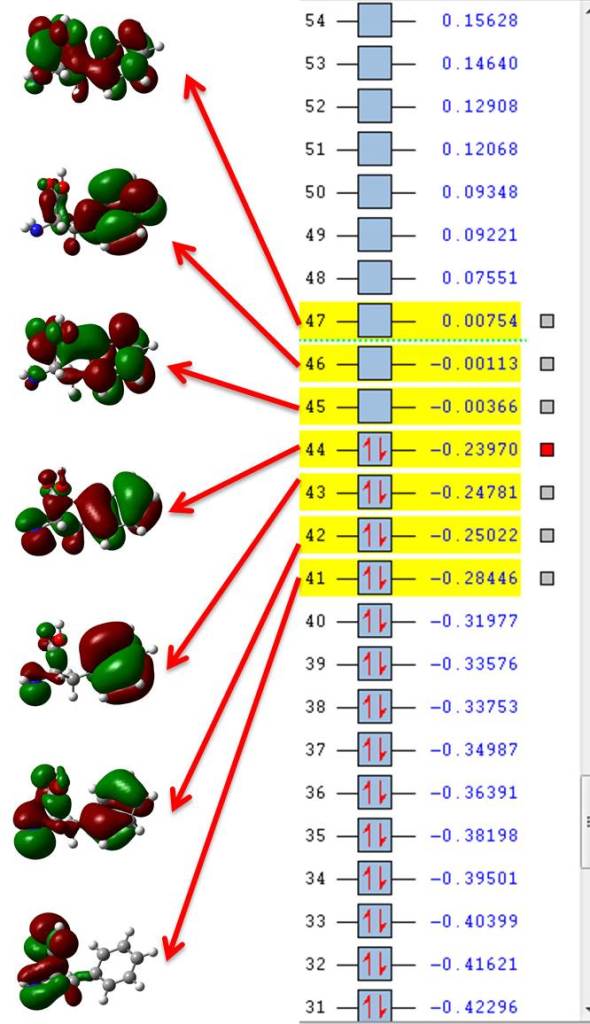
If we now open state10.fchk we see that the numbers at the side of the orbitals are not their energy but their occupation number particular to this state of interest, so we only need to plot those with highest occupations, in our example those are orbitals 44 and 45 (HOMO and LUMO) which have occupations = 0.81186; you may include 43 and 46 (HOMO-1 and LUMO+1, respectively) for a much more complete description (occupations = 0.18223) but we’re still dealing with 4 orbitals instead of 7.
The NTO transition 44 -> 45 is far easier to conceptualize than all the 10 combinations given in the canonical basis from the direct TD-DFT calculation. TD-DFT provides us with the correct transitions, NTOs just paint us a picture more readily available to the chemist mindset.
NOTE: for G09 revC and above, the %OldChk option is available, I haven’t personally tried it but using it to specify where the excitations are located and then write the NTOs of interest into a new chk file in the following way, thus eliminating the need of copying the original chk file for each state:
%OldChk=filename.chk
%chk=stateN.chk
NTOs are based on the Natural Hybrid orbitals vision by Löwdin and others, and it is said to be so straightforward that it has been re-discovered from time to time. Be that as it may, the NTO visualization provides a much clearer vision of the excitations occurring during a TD calculation.
Thanks for reading, stay home and stay safe during these harsh days everyone. Please share, rate and comment this and other posts.

Hello, I’m Brahim, beginner researcher in thoeritecal chemistry, I work with gaussian for three years ago, but I need someone to help and give a good way (orientation)? I have many new molecules no published, could you please help me for that?
Thank you. Your topic is clear. I confirm you that the %Oldchk command is going on as well. Please, could you show us how to perform, in Gaussview, a differential density map for the 10th excitation of your NTO example ? That is to say to show the redistribution of the electron density upon excitation (decrease and increase). Have a nice work.
Hello,
Thanks for confirming the functionality of %oldchk.
GaussView 5 and beyond is able to perform operations on cubes from the Surfaces window -> Cube Actions tab -> Add/Subtract/ two cubes.
So you first need to save the corresponding cubes from the NTO-containing fchk file with the cubegen utility. Once you generate them, load them and subtract them. I will try to write a step-by-step post on it.
Have a nice day
Hi Joaquin,
Thanks so much for your posts.
I’m using Orca 4.2.1 to do TD-DFT with TDA aproximation at the B97-3c composite model and it does calculation of NTO by default, it also has a clean organized output and it is free :-). My question : how that stands for open-shell neutral systems ? Can we still draw meaningful chemical view for transitions related to the NTO pictures corresponding to the excitation with the highest transition probability in open-shell neutral systems ?
Hola Edgardo,
Thanks for bringing ORCA to our attention, I should break some old habits and work other pieces of software I just need more time.
Yes, you can work it with open shell systems but you have to consider alpha and beta spin densities separately, beware of the occupation numbers.
I hope this helps!
Hola Joaquin,
Thanks for your answer.
True, breaking habits is never easy. This pandemic made me spend more time at the computer, found out that G09-ORCA oscillator strength is much higher than I anticipated. Operation is actually similar to G09 with keywords text input, really nice output files, helpful active forum, many options to generate cube files, visualizers include GaussView and free ones such as iQmol, Gabedit, Avogadro, Chimera etc
I’m just starting to work with ORCA, mainly because it has several brand new DFT methods better suited to exited states and transitions in organo-metallic complex. That brought me into NTOs and your cool blog.
One thing I noticed with the usual canonical molecular orbital isodensity pictures, is that their shapes can be highly dependent on the basis set used, I mean different pictures and localization for the same molecule. When that is the case their usefulness in providing any ‘chemical meaning’ is highly questionable.
So far I only tried with a handful of compounds, found NTO much better in providing a more meaningful (localized) picture for particle-hole transitions.
In your experience, are the NTO pictures consistent for a given molecule, less dependent than canonical MOs on the basis sets and DFT methods ?
Hi Joaquin,
Thanks for your very useful posts. I want to ask you about the cases when, after an NTO calculation, several hole-particle pairs are obtained, and some of them have important occupations, let’s say 0.6 and 0.3. Is it possible to have an even better visualization by just performing a weighed sum of the resultant hole cubes and then, separately, the particle cubes? I have tried this and got a consistent result, but I am not sure if it is correct.
Hola Pablo
It sounds right, I mean in the end there is nothing that prevents you from adding several NTO cubes into one and in the end each molecular orbital is the linear combination of other orbitals. I may be forgetting something here but I think there isn’t any fundamental prohibition to perform this linear combination (I’m assuming your weighing coefficients are the relative occupations). The only thing that would be fundamentally wrong would be to sum hole and particle NTOs
Gracias por leernos!
This is an interesting question I have been wondering about too, trying to get a better idea of how to interpret these NTOs. There are 2 linear combos I was thinking of, but not sure how to pick one or the other, one sounds like what you describe maybe:
h_j are holes, e_j are particles, n_j are the eigenvalues
Psi_1(x_e, x_h) = (n_1*h_1 + n_2*h_2 + …. ) * (n_1*e_1 + n_2*e_2 + …)
or
Psi_2(x_e, x_h) = n_1(h_1*e_1) + n_2(h_2*e_2) + ….
I am still grappling with what the NTOs ‘mean’ etc., would love to discuss more.
Hello professor Barroso,
Thank you for this instructional post! I really appreciate it!
I had a quick question, I performed a a TD-DFT with nbo, and it was nice to see the localized picture of the orbitals.
Afterwards I performed a NTO analysis on excited state #3, my questions are as follows:
1) Does the numbering of orbitals in NTO the same as NBO.For instance is orbital number 254 in NBO (for all excited state) the same as the orbital labeled 254 after the NTO for the third excited state?
2) Also, in the NTO, I see my two highest occupation orbitals (occ and virtual) are one where the picture is very deolocalized over the entire molecule and the virtual orbital is centered on the metal, can I interpret this as an increase in electron density at the metal in the given transition?
Dear Kekeedme
I’m glad you found it useful.
The short answer for both your questions is yes 🙂 once NBO labels an orbital it sticks to that numbering scheme throughout any other analysis provided you’re using the same input/checkpoint file, so as long as you don’t start your calculation from scratch (and make a mistake then) you should be fine considering the same numbering scheme.
NTOs are usually localized but sometimes they don’t look very much so, unfortunately. In the case you’re describing it looks like you have a charge transfer case.
Thank you for reading.
Thanks for this great write-up, Joaquin. It’s very clear and helpful for understanding the principles (and practicals) of NTO analysis. 🙂
Hello Doctor, your blog has very useful information for those of us who are starting with this.
Is it possible that you have or can provide me some example on how to calculate Singlet-Triplet Split energy (S1 to T1) in gaussian? I mean, about the sequence to follow, or some example of the inputs.
Thanks in advance
Hello professor Barroso,
I came here from a former question about calculating the orbitals of excited states (https://www.researchgate.net/post/How-can-I-calculate-the-natural-transition-orbitals-analysis-NTO-and-visualize-it). I followed the input “#p B3LYP/6-31G(d,p) Geom=AllCheck Guess=(Read,Only) Density=(Check,Transition=N)”, as well as “% oldchk” and “% chk”. However, it still can’t solve the “This type of calculation cannot be archived” problem.
When I did not add “% oldchk”, the .log file wrote “This type of calculation cannot be archived”.
When I added the “% oldchk”, the .log file indicated that it just copyed the information in old chk file to the new one.
Did you find the answer to your problem? I am having exactly the same one
You may want to check:
Sorry for the automatic and cyclic response. You should then just copy chk files. Make sure you’re not asking for opt and maybe get rid of ONLY in Guess.
You may want to check:
Sorry for the automatic and cyclic response. You should then just copy chk files. Make sure you’re not asking for opt and maybe get rid of ONLY in Guess.
Hello Dr., please the same question asked by Erick Alfonso. Many tanks! Your boog is so grea!t
Hello sir, I repeated the same calculation for phenylalanine molecule at the same level of theory using G09 program. But the obtained results from the TD-DFT calculation is quite different mostly if I compare the oscillator strengths. In my case the oscillator strength of S10 state is 0.135 which contains 7 transitions while here it is 0.36 which contains 8 transitions. Please help me in finding out such type of discrepancies. Thank you.
Thank you for the excellent tutorial, it is helpful for beginners like myself. I want to make sure I am calculating what I think I am calculating using G16 and I was hoping you could help. I have run a TDDFT calculation, which has in the log file:
Excited State 1: Triplet-A 0.3654 eV 3392.76 nm f=0.0000
=2.0001251 ->1256 -0.13656
1252 ->1255 -0.14126
1253 ->1254 0.68058
1253 <-1254 0.10520
This state for optimization and/or second-order correction.
Total Energy, E(TD-HF/TD-DFT) = -15898.6618005
Copying the excited state density for this state as the 1-particle RhoCI density.
Excited State 2: Singlet-A 0.5235 eV 2368.52 nm f=0.0378
=0.0001252 ->1254 -0.13007
1253 ->1254 0.67648
1253 ->1255 0.13450
Excited State 3: Triplet-A 0.6423 eV 1930.35 nm f=0.0000
=2.0001252 ->1254 0.54980
1253 ->1255 -0.42366
….etc.
If I then run NTO analysis using
#b3lyp/6-31g Geom=AllCheck Guess=(Read,Only) Density=(Check,Transition=2) Pop=(Minimal,NTO,SaveNTO)
Does this perform NTO on that Excited State 2 listed in the log? Or on the second singlet excited state in the list (not shown)? The ground state is singlet. Thank you.
Not sure why that’s struck-through, my apologies.
YEs, it precisely does it on state 2. I hope this helps!
Respected sir,
I can find the energy gap from singlet state of homo lumo. What should I do to find the energy gap of doublet spin since there are four corresponding columns in MO editor.
Thank you for this very useful blog !
If possible, a question I have in mind related to what I’m doing, and that’s trying the idea of using Density=transition(N,M). Is it possible to get the NTO for a transition happening between two excited states? I tried using it, but for some reason I don’t get any useful output, as it just gives me the same NTOs calculated for a ground-excited transition. Thanks !
very good information. but I had seen in some papers they are mentioning the occupied orbital as a hole and the unoccupied orbital as a particle( which is normal second quantization formalism). you mentioned it in a reverse way. Can u clarify this thing?
It would be helpful to see a COMPLETE gjf file, even for a relatively simple molecule.