
It is with great pleasure that I’d like to announce the thesis defense of Guillermo “Memo” Caballero and Howard Diaz who in past days became the second and third students, respectively, to get their B.Sc. degrees with theses completed at our lab. I want to publicly thank them for their hard work which hasn’t only contributed with a thesis to our library but will soon contribute with research papers to our count.
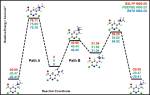
Guillermo “Memo” Caballero worked on the calculation of a reaction mechanism that cannot happen. He started as a synthetic chemist and when he hit a wall at the lab he thought computational chemistry might help him get synthesis on the right direction. He has proven now that the aromatization process of a substituted glutarimide into the corresponding pyridine can only proceed only if substituents with a very strong electron withdrawing effect are used. For two reaction mechanisms proposed, both of them intramolecular rearrangements and only one of them concerted, the calculated energy barriers to reach for the corresponding transition states (QST2 and QST3 methods used) are higher than a pyrolitic decomposition. Memo found also that the delocalization of the pi electron system and its extent goes a long way into the stabilization of the non-aromatic analogue. At first we wanted to treat this problem as a tautomeric equilibrium but since we cannot observe the aromatic tautomer there is no equilibrium and hence no tautomerism. We are still thinking how to name this correspondence between the two compounds when we submit the corresponding paper. It must be said that Guillermo graduated with the highest honors in a most deserved way.


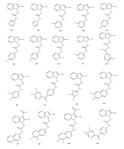
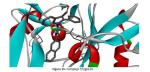
Howard Diaz worked on the design of molecular blockers for the entrance process of the HIV-1 virus into lymphocytes through the GP120 protein. Six known blockers based on phenyl-indoyl-urea were assessed through docking, the binding site of the GP120 protein was described in terms of the interactions formed with each on these compounds and that served as the basis for what in the end came up to be a 36 compound library of blockers, whose structures were first optimized at the B3LYP/6-31G** level of theory. All the 42 blockers were docked in the binding site of the protein and a thorough conformational search was performed. From this set, lead compounds were selected in terms of their binding energies (first calculated heuristically) and further studied at the Density Functional Theory, B97D/cc-pVTZ in order to study the electronic structure of the blocker when interacting with a selection of residues at the binding site. Interaction energies calculated at the quantum level are consistent with the complex formation but since we had to cut the protein to only a few residues little correlation is found with the first calculation; this is fine and still publishable, I just wish we had a more seamless transition between heuristics and quantum chemical calculations. Wiberg indexes were very low, as consistent with a hydrophobic cavity, and delocalization energies calculated with second order perturbation theory analysis on the Natural Bond Orbitals revealed that the two most important interactions are C-H…π and Cl…π, these two were selected as key parameters in our design of new drugs for preventing the HIV-1 virus to bind lymphocytes-T; now we only need to have them synthesized and tested (anyone interested?).


Thank you guys for all your hard work, it has truly payed off. I’m completely certain that no matter what you do and where you go you will be very successful in your careers and I wish you nothing but the very best. This lab’s doors will always remain open for you.


Dear Dr. Barrosso,
I am very happy to see your post on computational chemistry. I do a small part of work in this field.It is very interesting field.
cher Dr Joaquinbonjour, mes félicitations à tous les membres de votre laboratoire et spécialement à *Guillermo “Memo” Caballero et Howard Diaz* je suis un doctorant chimiste Algérien entre le mure du laboratoire de synthèse et la chimie computationnelle et théorique. pour ce la j`ai besoin d`aide en première étape SVP des copies des thèses soutenus de *Guillermo “Memo” Caballero et Howard Diaz*Cordialement
Mercie beaucup, I have no French knowledge, sorry.
I think its an interesting idea, I will talk to Guillermo about it. In the mean time there is a presentation on my slideshare profile about his work in case you want to search for it. We hope we manage to have his manuscript published soon this year.
Bon jour!
No doubt that the world is so small.
Congratulations on this great achievement, I met these guys in the RMFQT 2014 in Morelia, I noticed that they are a good young researchers. I have been a follower of your blog for a few months, I’ve even done some questions.I hope to keep in contact with you and your work team.
Regards from Chihuahua.
Hi,
I am new to computational chemistry and my project now involves the use DFT calculations to find my products transition state/intermediate. The reaction involves a hydride shift followed by cyclisation to get to the final product. I am trying to find the best protecting group for my nitrogen atom and will be looking at several different protecting groups. Do you have a suggestions of literature and test files I could use to get my head around these calculations? Any advice would be much appreciated!
Many Thanks
There are several tutorials online for transition state searches. If you have Gaussian available you can find a copy of “Exploring Chemistry with Electronic Structure Methods” by Frisch and Frisch, therein you can find some examples with the corresponding input files.
Try the following three posts from this blog:
I hope this helps.
Hi,
I am new to computational chemistry and my project now involves the use DFT calculations to find my best sensizer to use in DSSC I am trying to find the best compound.and i want group or team research to work with and have thier advices Do you have a suggestions of that
and first i want to interpret NBO terms such as BD CR RAY.. etc and how to interpret them as pi and sigma and pi* and sigma*. Any advice would be much appreciated!
Hello Mohammed,
For a quick overview of how to interpret NBO calculations, please check my blog and the following entries:
https://joaquinbarroso.com/category/nbo/
Now, for the first part of your question, you’re describing a massive project. I suggest that if you’re new to comp.chem. you search for a collaboration with a well established group who already perform the calculations you’re trying to achieve, that way they can guide you more precisely.
Have a nice day